Этот пост очень длинный, так как содержит мои полные письменные показания для голландского суда. Структура поста следующая:
Информация по делу и ссылки на видео на YouTube-канале RechtOprecht.
Освещение дела в новостях
клеветническая кампания
Краткое изложение моих показаний
Полное свидетельство
1. Предыстория дела:
Для тех, кто не знаком с этим делом, это короткое видеообращение от Питера Стассена, единственного оставшегося адвоката по этому делу. Его коллега, Арно ван Кессель, был заключен в тюрьму по [сфабрикованным] обвинениям в терроризме и лишен адвокатской лицензии:
2. Освещение в новостях:
В недавней статье издания The Defender содержится отличное резюме дела и событий, произошедших на данный момент:
По мнению экспертов, вакцины от COVID-19 «неотличимы от биологического оружия».
В ходе пресс-конференции Стассен также отметил свои усилия по тому, чтобы голландские суды приняли показания его экспертов, данные лично. Он сказал, что свидетели намерены представить доказательства, свидетельствующие о том, что вакцины от COVID-19:
Они «неотличимы от биологического оружия».
Не предоставляют никаких преимуществ для здоровья.
Они «не являются ни безопасными, ни эффективными».
В США они были выпущены в рамках разрешения на экстренное использование , «правового статуса, который снимает с FDA (Управление по контролю за продуктами и лекарствами США) действие закона о фармацевтической продукции и защите прав потребителей».
Они "по замыслу врача предназначены для причинения вреда, описанного в инструкции по применению и в отчетах как „ побочные эффекты “", включая " внезапную смерть , сердечную недостаточность , рак и самые ужасные заболевания".
Являются «ключевым компонентом» « Великой перезагрузки », «военного проекта, в котором НАТО играет значительную роль ».
Это репортаж об этом деле из голландской газеты (нажмите на кнопку перевода на английский): « Суд постановил, что ответчики должны явиться на слушание лично ».
Это моё короткое видеосвидетельство, в котором я кратко излагаю содержание своего письменного заявления (полный текст, за исключением приложения 1G, приведён ниже):
На этом YouTube-канале доступны видеообзоры от Майка Йидона, Кэтрин Уотт, Кэтрин Остин Фиттс и Джо Сансоне, а также другие видеоинтервью .
3. Клеветническая кампания:
Обычно я стараюсь избегать ненужной чепухи из социальных сетей в своих статьях, но эта информация очень важна и актуальна, поэтому её необходимо обсудить. Если вы являетесь подписчиком Jikky Leaks (он же Mouse Piss) и связанной с ним «армии мышей» (она же Moussad) на X, или подписчиком доктора Ах Кана Сайеда (тот же человек (люди), что и Jikky на X), или OpenVAET (он же «canceledmouse» на X) и многих других лжецов, анонимных или названных троллей с логотипами мышей в профиле — будьте осторожны! Это сеть агентов, финансируемых военными, разведывательными и частными интересами, и их работа заключается в том, чтобы разжигать споры и «увеличивать двусмысленность» (термин разведки), генерируя множество, казалось бы, релевантного контента, но цель состоит в том, чтобы запутать и увести от того, что действительно важно в расследовании. Например, увести от правовых/нормативных рамок/военного права в бесконечные и непродуктивные дебаты о «золотом стандарте науки». Это часто называют «агентами хаоса» — хаос или дезорганизация в противоположность космосу, структуре и ясности. Эти операции также направлены на очернение людей, которые находятся на правильном пути в отношении борьбы с COVID-19 и, что наиболее важно, добиваются ощутимого прогресса. Упомянутые выше агенты притворяются, что находятся на правильной стороне, публикуя множество технически обоснованных материалов и критикуя вакцины. Однако они настаивают на том, чтобы все преследовали только крупные фармацевтические компании и оставались в рамках дискуссии о «золотом стандарте науки». Как известно моим читателям, настоящее преступление, совершенное во время пандемии COVID-19, не имеет ничего общего с наукой. Оно связано с скоординированной на глобальном уровне военной атакой на общество , оплачиваемой из военных бюджетов/контрактов, с заказом веществ и протоколов, которые неотличимы от оружия, поставляемого частными компаниями-сообщниками из крупных медицинских/фармацевтических компаний.
Вот немедленная ложь Джикки в ответ на статью в издании The Defender, ссылку на которую я привела выше:
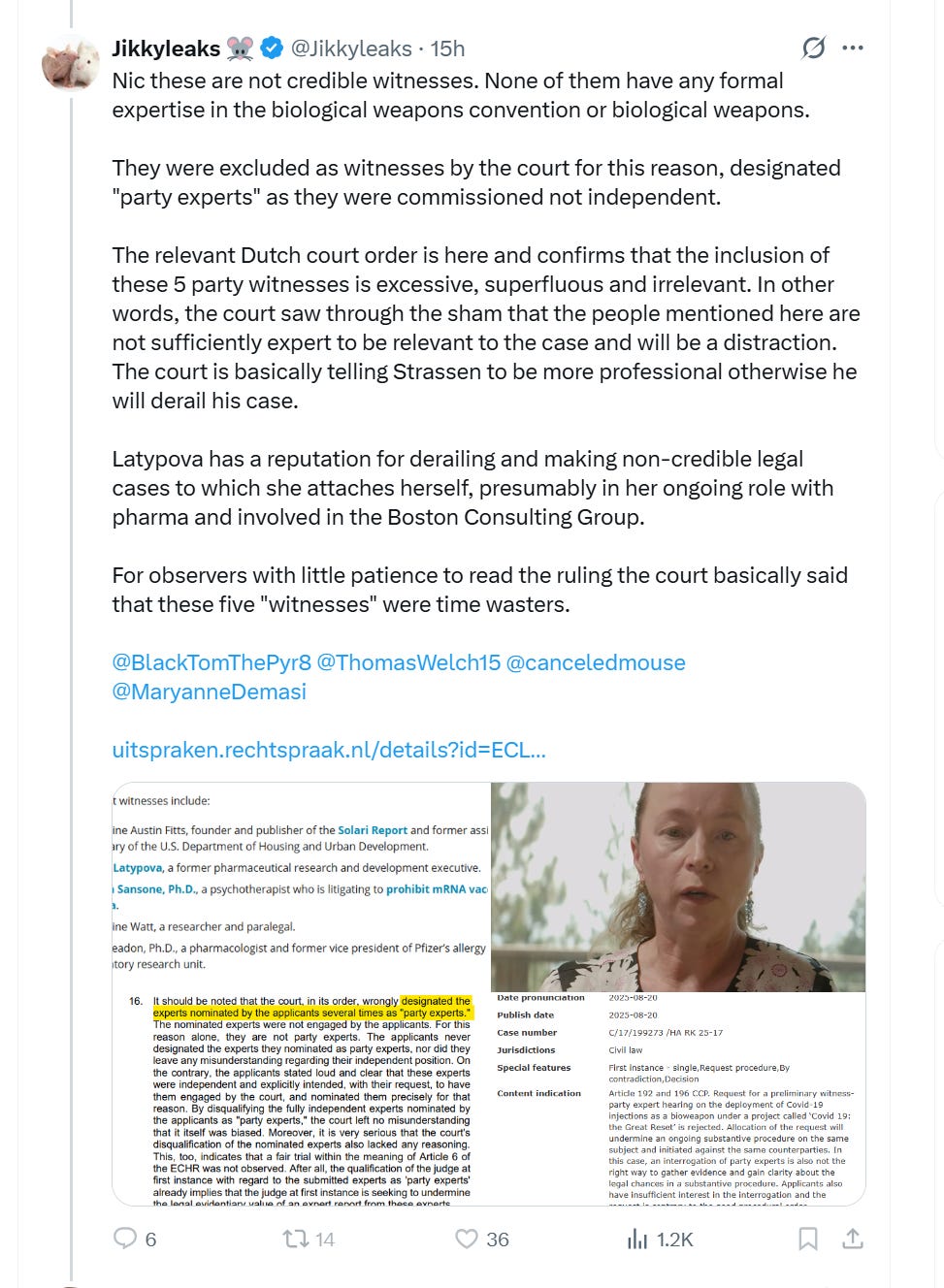
Обратите внимание, что всё, что он пишет обо мне, — ложь:
Он говорит, что я работаю в фармацевтической компании — это ложь, я ушел из фармацевтической отрасли почти 10 лет назад.
Он говорит, что я связан с Boston Consulting Group — это ложь. У меня нет никаких связей с BCG, я никогда там не работал и даже не участвовал ни в одном контракте с ними.
Он утверждает, что я «срываю» судебные процессы, и упоминает дело Кирка Мура! Это просто смешно! Мои и материалы Кэтрин Уотт сыграли решающую роль в прекращении этого дела Пэм Бонди. Так что, если он думает, что я «срываю» дела, следует спросить — срываю ли я их ДЛЯ КОГО? Очевидно, это показывает, на кого работает Джикки/Саиди, да, именно на этих людей я действительно срываю дела.
Вот коллаж из нескольких имен Джикки Ликс, также известной как «Мышиная моча»:

А вот и настоящая личность Джикки — психопат, фальшивый «доктор», специализирующийся на издевательствах и насилии над женщинами. Однако он не настоящий врач, он почти 100% своего времени проводит в интернете, занимаясь троллингом для тех, кто ему за это платит. И это хорошая новость, потому что он больше не может никому причинить физический вред:

Хватит уже этой чепухи.
Мои письменные экспертные показания, представленные суду 15 сентября 2025 года:
4. Краткое изложение
Область применения и экспертное мнение:
На основании анализа основных нормативных документов, просочившихся в прессу файлов Pfizer по химическому контролю производства (CMC), соответствующего законодательства США и ЕС, а также другой общедоступной документации, я, как эксперт, пришел к выводу, что инъекции мРНК COVID-19 применялись в соответствии с военными правилами «медицинского противодействия», которые обошли стандартные фармацевтические меры безопасности, что делает их юридически и функционально неотличимыми от потенциального биохимического оружия.
1. Двойное назначение платформ мРНК/ДНК
Установлено обозначение «двойного назначения» – по меньшей мере с 1997 года американские советники по вопросам обороны (группа JASON), а позже и Национальные академии США, включают платформы генной терапии, в том числе системы мРНК на основе липидных наночастиц (ЛНП), используемые в качестве вакцин, в список технологий, которые могут быть использованы в качестве оружия (например, путем доставки токсинов, онкогенов или иммунодепрессивных микроРНК).
Легкость использования в качестве оружия – Те же самые свойства, которые делают мРНК привлекательной для терапии (проникновение в клетки, высокая экспрессия), делают ее привлекательной и для враждебного применения; даже фрагментированная РНК (shRNA, miRNA) может нарушать регуляцию экспрессии генов хозяина, не кодируя белки.
2. «Обман покупателей»: потребителей во всем мире ввели в заблуждение относительно правового статуса товаров, связанных с COVID-19, как средств противодействия – лекарств, используемых в немедицинских целях, то есть в качестве оружия.
Для продуктов, содержащих мРНК COVID-19, были отменены обычно действующие меры защиты потребителей:
Правила проведения клинических исследований новых лекарственных препаратов/надзор этического комитета: препараты, ранее разработанные в качестве генной терапии, были переклассифицированы как «вакцины/средства противодействия» исключительно на основании заявленного предполагаемого применения; требования к безопасности, фармакологии, канцерогенности и мутагенности были отменены; объявление чрезвычайных ситуаций и прекращение клинических исследований средств противодействия лишили возможности применять все фармацевтические правила.
Инспекции в соответствии с действующими правилами надлежащей производственной практики (cGMP): Закон PREP (США) и Регламент ЕС о чрезвычайной поддержке 2016/369 (с поправками 2020 г.) позволяют министру/Комиссиям полностью отказаться от соблюдения cGMP во время объявленной чрезвычайной ситуации, связанной с химическим, биологическим, радиологическим и ядерным оружием.
Правила импорта-экспорта и ответственность производителей: отменены в рамках соглашений ЕС о поставках с фармацевтическими компаниями. Полная защита фармацевтических компаний от любых травм или смерти, вызванных небезопасной продукцией, за исключением случаев узко определенного «умышленного нарушения» – аналогично Закону США о предотвращении рисков для здоровья (PREP Act) в контрактах ЕС.
Независимые испытания партий продукции в ЕС: Соглашение о взаимном признании FDA-EMA (полностью вступившее в силу в июле 2019 года) позволяет квалифицированным специалистам ЕС принимать данные о партиях продукции, полученные в США, без предварительного ознакомления с ними.
Инспекции предприятий проводились каждые два года (21 CFR 600.21): в апреле 2019 года правила FDA отменили частоту инспекций и штрафные санкции; затем запреты на поездки, связанные с COVID-19, полностью приостановили инспекции.
Документальные доказательства несоблюдения надлежащей производственной практики и фальсификации продукции (потенциальное использование в качестве оружия):
Утечки информации от EMA (ноябрь 2020 г.) – около 1000 страниц содержат основные возражения: (i) отсутствие упаковки cGMP; (ii) содержание интактной мРНК ≤50 % (спецификация незаметно снижена с ≥70 %); (iii) отсутствие валидации аналитического метода; (iv) непроверенные изменения в производстве.
В отчете FDA Form 483 за 2022 год , подготовленном компанией Rentschler, зафиксированы нарушения надлежащей производственной практики (cGMP), подтвержденные ранее вынесенными предупреждениями. Никаких мер принудительного характера не принято.
Правовая защита – статья 21USC§360bbb‑3a(c) прямо указывает, что даже если продукт «фальсифицирован или имеет неправильную маркировку», он не может рассматриваться как таковой после того, как он был обозначен как контрмера в рамках экстренного разрешения на использование. Аналогичные условия, освобождающие от регулирования и ответственности, были реализованы в ЕС посредством хищнических договоров закупок.
3. Доказательства того, что меры реагирования на COVID-19 не были мерами в области общественного здравоохранения, а представляли собой засекреченную военную операцию с использованием медикаментов в качестве оружия.
Все средства противодействия COVID-19, включая биохимические вещества, продаваемые как «безопасные и эффективные вакцины», были заказаны Министерством обороны в рамках «демонстрации крупномасштабного производства» по контрактам, заключенным в соответствии с Законом о других транзакциях. Согласно отчетам помощника министра здравоохранения и социальных служб США по вопросам готовности и реагирования в рамках операции Warp Speed, Министерство обороны США (DoD) заказало и контролировало разработку, производство и распространение всех средств противодействия.
Досье по COVID-19 (Приложение 1) представляет собой подборку данных из многих стран и регионов мира, демонстрирующих, что:
COVID-19 не был событием, касающимся общественного здравоохранения, хотя и представлялся таковым населению мира. Это была глобальная операция, скоординированная посредством государственно-частных разведывательных и военных альянсов, а также с применением законов, разработанных для атак с использованием химического, биологического, радиологического и ядерного оружия (ХБРЯ).
Досье содержит информацию о координации действий военных и разведывательных служб в рамках мер биологической защиты от COVID-19 в США, Великобритании, Австралии, Канаде, Нидерландах, Германии, Италии и многих других странах. Для максимально возможного числа стран в досье указаны военные/разведывательные органы, ответственные за меры реагирования на COVID-19 в их странах; даты объявления чрезвычайного положения в каждой стране; связанные с военными/разведывательными службами и органами, отвечающими за цензуру/пропаганду; а также высокопоставленные лица, занимавшие руководящие должности в военных/разведывательных органах, которые, как известно или сообщается, занимали руководящие позиции в рамках мер реагирования. В досье также указаны связи с глобальными руководящими органами, включая ЕС и ООН/ВОЗ, через которые координировались меры реагирования, и приведен список военно-разведывательных/биологических альянсов, которые обеспечили многонациональные рамки для реагирования на биотеррористическую атаку/атаку с применением биологического оружия.
4. Инъекции мРНК COVID-19 неотличимы от биохимического оружия.
Инъекции мРНК COVID-19 соответствуют законодательному определению «биологического продукта», используемого в качестве средства противодействия немедицинским, неразрешенным целям, и одновременно освобождаются от действия законодательства о безопасности лекарственных средств и ответственности производителей.
Потенциальная квалификация в качестве «биологического оружия» согласно статье 175 и далее раздела 18 Свода законов США (владение или распространение любого вещества, «не оправданное профилактическими или защитными целями») с учетом доказательств систематического фальсификации и неправильной маркировки.
Предсказуемость вреда – В нормативных документах по генной терапии и обширной научной литературе в качестве известных рисков указаны инсерционная мутагенеза, аутоиммунные реакции и персистирующая экспрессия; эти риски не были ни проверены, ни раскрыты. Отсутствие соответствия стандартам надлежащей производственной практики (cGMP), свидетельства фальсификации и загрязнения, а также значительные пробелы в характеристиках производственного процесса на момент глобального запуска демонстрируют вопиющее безразличие государства и органов здравоохранения, преднамеренно подвергающих миллионы людей риску смерти и серьезных травм.
Ответственность за действия третьих лиц – Ответчики знали или должны были знать, что не существовало законного разрешения на применение инъекций мРНК COVID-19 в фармацевтической промышленности, и что миллионы людей подвергались предсказуемому вреду. Тем не менее, они продолжали лгать общественности и применять принудительные меры для увеличения числа вакцинированных.
Выводы:
Научный аспект – Присущая платформам LNP-мРНК опасность двойного применения требует самого тщательного контроля со стороны производителей и регулирующих органов; однако наблюдается обратная ситуация.
Регулирование – Благодаря согласованной глобальной стратегии (Закон о профилактике и профилактике заболеваний, контрмеры, чрезвычайные положения ЕС, соглашения о взаимном признании) действие обычного законодательства о безопасности лекарственных средств было приостановлено, что позволило бесконтрольно фальсифицировать продукцию.
Судебно-медицинская экспертиза – Утечка файлов EMA и последующие результаты инспекций FDA подтверждают объективные производственные сбои, соответствующие путям создания биологического оружия, описанным в американской литературе по биологической защите.
Правовые аспекты – Согласно законодательству США и международному праву, продукция, выпускаемая под видом лекарственного средства, но прошедшая функциональные испытания на предмет пригодности к применению в качестве биохимического оружия, влечет за собой потенциальную уголовную ответственность для всех участников цепочки поставок.
Документ подготовлен для предоставления в качестве основного пояснения; подробные ссылки, приложения и резюме предоставляются по запросу.
5. Полное свидетельство:
1. Двойное назначение платформ мРНК/ДНК
Биологическое и биохимическое оружие – это как природные, так и созданные человеком материалы, предназначенные для вызывания заболеваний путем введения токсинов и микроорганизмов. Способ применения биохимического оружия зависит от самого вещества, его подготовки, стойкости и способа введения. Нападающие могут распространять эти вещества в виде аэрозолей или через пищу и воду [1] . Помимо внешнего распространения таких веществ, в литературе, посвященной биохимическому оружию и терроризму, описывается введение биохимических агентов и другими способами. В частности, в качестве возможных векторов нападения описывается использование в потребительских товарах, лекарствах или даже кормах для домашних животных и других продуктах [2] .
В политике, дипломатии и экспортном контроле «двойное назначение» относится к технологиям, которые могут использоваться как в мирных, так и в военных целях. Технология мРНК/ДНК, включая варианты в виде инъекционных лекарств или вакцин, давно признана технологией двойного назначения, потенциально пригодной для создания оружия [3] , [4] , [5] . «Дилемма двойного назначения» впервые была отмечена с открытием процесса синтеза и массового производства аммиака, который произвел революцию в сельском хозяйстве благодаря современным удобрениям, но также привел к созданию химического оружия во время Первой мировой войны. Эта дилемма давно признана в химии и физике, что привело к принятию международных конвенций и договоров, включая Конвенцию о химическом оружии и Договор о нераспространении ядерного оружия.
В 1997 году группа JASON [6] , консультативный комитет при президенте США по научным вопросам, касающимся военных технологий, выявила потенциал генно-модифицированных патогенов в следующих шести группах:
Наивысшая форма
Нижняя часть формы
Бинарное биологическое оружие
Гены, созданные дизайнером
Генная терапия как оружие
Скрытые вирусы
болезни, связанные со сменой хозяина
Болезни, созданные дизайнерами
На сегодняшний день системы информирования и контроля для выявления и предотвращения потенциальной подрывной деятельности и злоупотреблений остаются крайне неэффективными. Несмотря на очевидную угрозу, исходящую от передовых продуктов синтетической биологии в случае их использования в качестве оружия, сегодня практически отсутствуют какие-либо меры контроля, за исключением нескольких руководящих документов для ученых и исследовательских учреждений. Эти требования к самоотчетности в значительной степени игнорируются, и, как показывают гранты NIH Уханьскому институту вирусологии, исследования, которые могут вызвать возражения, переносятся за границу.
1.1. Методы создания оружия на основе мРНК/ДНК-технологий и продуктов:
В принципе, любой лекарственный препарат или инъекционный медицинский продукт может быть использован в качестве оружия, то есть в качестве яда вместо лекарства. Это связано с тем, что потребитель или медицинский работник не могут напрямую оценить продукт и его состав и должны полагаться на регулирующие органы, обеспечивающие соблюдение фармацевтического законодательства/правил, для гарантии соответствия и безопасности продукта на всех этапах его производства, поставок и распространения. Фармацевтические вещества содержат потенциально опасные химические соединения, и, как правило, разница между лекарством и ядом/смертельным оружием заключается в точной дозировке, которую фармацевтические правила призваны строго контролировать. В качестве примера рассмотрим опиоиды, используемые в законных целях — для обезболивания, против их потенциальной смертельной передозировки. Неправильно маркированный опиоидный препарат с неверными указаниями дозировки будет примером лекарственного средства, используемого в качестве оружия.
Согласно литературе по биологическому оружию, создание оружия на основе генной терапии/мРНК-технологий или других платформ ДНК/РНК может быть осуществлено различными способами. Синтетическая мРНК — это большая молекула (~3000+ пар оснований). Она нестабильна и хрупка, распадаясь на более мелкие сегменты в процессе производства, хранения и транспортировки. Было показано, что сегменты РНК, такие как короткие шпилечные РНК (shRNA) или микроРНК (miRNA), могут быть использованы в качестве оружия, и для этого не требуется точная модификация, а также отсутствие необходимости «кодировать» что-либо конкретное.
Следующий абзац находится в главе 6 учебника «Биозащита в эпоху синтетической биологии» 2018 года [7] :
«Малые РНК являются примером функциональной генетической информации, которая может передаваться горизонтально. Малые РНК, хотя и не являются модификацией генома как таковой, важны, поскольку они могут оказаться способными изменять экспрессию генов и вызывать фенотипические изменения. Большое количество исследований малых интерферирующих РНК (siRNA), коротких шпилечных РНК (shRNA), микроРНК (miRNA) (Zhang et al., 2007; Huang et al., 2008) и других библиотек малых РНК в различных видах и клетках разных видов, включая человека, предоставляет потенциальную дорожную карту того, какие последовательности могут приводить к каким патологическим состояниям или к модуляции защиты от болезней. {…}»
Одна из причин, по которой доставка РНК потенциально представляет собой реальную биологическую угрозу, заключается в том, что даже небольшое первоначальное изменение экспрессии генов (например, изменения экспрессии генов, обычно вызываемые микроРНК) может значительно изменить вероятность первоначального изменения клеток. Даже небольшие количества целевой РНК сами по себе не изменят геном, но могут позволить или побудить клетки начать процесс самотрансформации в опухоли, о чем свидетельствует тот факт, что уже обнаружено большое количество проонкогенных микроРНК (O'Bryan et al., 2017). Помимо РНК, продуцируемых вирусами, бактерии продуцируют многочисленные малые регуляторные РНК; их введение в эндогенный микробиом может привести к дисбиозу. Более крупные мРНК также могут доставляться с помощью липосом и наночастиц или с помощью стратегий репликации РНК, разрабатываемых для производства вакцин (см. главу 8, «Быстрая разработка самоамплифицирующихся мРНК-вакцин»); Эти методы потенциально могут быть использованы для экспрессии вредных веществ, таких как токсины или онкогены, аналогично угрозам, связанным с ДНК-векторами.
Очевидно, что как короткие, так и более длинные последовательности РНК могут быть введены в организм человека в злонамеренных целях, включая развитие рака, дисбиоза, подавление иммунной системы и повреждение органов. Это может быть достигнуто как с включением, так и без включения кода РНК, предназначенной для использования в качестве оружия, в геном хозяина, а также с помощью последовательностей РНК, которые не должны кодировать какой-либо белок.
Кроме того, в том же учебнике утверждается, что вакцины рассматриваются как потенциальное средство для создания оружия:
« Более крупные мРНК также можно доставлять с помощью липосом и наночастиц или посредством стратегий репликации РНК, разрабатываемых для производства вакцин [имеется в виду платформа самоамплифицирующихся мРНК-вакцин] … эти методы потенциально могут быть использованы для экспрессии вредных веществ, таких как токсины или онкогены».
Таким образом, мРНК/ДНК-препараты, продаваемые как «вакцины», в принципе, подвержены фальсификации, как преднамеренной, так и из-за отсутствия надлежащего контроля производственного процесса и определения чистоты. Технические возможности, необходимые для надежной характеристики и контроля этих веществ на этапах производства, распространения и применения, сегодня находятся на начальной стадии развития и не были на регулярной основе созданы производителями и регулирующими органами в 2020-2021 годах, когда эти вещества были массово внедрены по всему миру.
2. «Обман покупателей»: потребителей во всем мире ввели в заблуждение относительно правового статуса товаров, связанных с COVID-19, как средств противодействия – лекарств, используемых в немедицинских целях, то есть в качестве оружия.
2.1. Краткое изложение правовых требований законодательства США и ЕС в отношении продвижения фармацевтических препаратов как «безопасных и эффективных».
Как в Соединенных Штатах, так и в Европейском Союзе фармацевтическое регулирование призвано обеспечить безопасность, эффективность и высокое качество лекарственных препаратов до того, как они поступят в продажу. Хотя между системами США и ЕС существует много сходств, особенно в их опоре на научные данные, каждая из них имеет свои собственные регулирующие органы и процедуры оценки лекарственных препаратов и утверждения заявлений о безопасности и эффективности.
В Соединенных Штатах Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) через свой Центр оценки и исследований лекарственных средств (CDER) и Центр оценки и исследований биологических препаратов (CBER) является основным органом, ответственным за утверждение фармацевтических продуктов. Прежде чем новый лекарственный препарат сможет быть выведен на рынок, спонсор должен подать заявку на регистрацию нового лекарственного препарата (NDA) или заявку на получение лицензии на биологический препарат (BLA), которая включает убедительные данные доклинических исследований и клинических испытаний, демонстрирующие безопасность и эффективность продукта для его предполагаемого применения. Процесс клинических испытаний требует наличия у продукта статуса исследуемого препарата в рамках разрешения на проведение клинических испытаний и проходит структурированную последовательность фаз I–III с формальным надзором со стороны независимого наблюдательного совета по исследованиям (IRB), обеспечивающего защиту участников исследования и выполнение надлежащих процедур информированного согласия. Только после этого тщательного процесса, завершающегося положительной оценкой соотношения риска и пользы, могут быть сделаны заявления о безопасности и эффективности. FDA также проверяет соответствие производителя действующим правилам надлежащей производственной и клинической практики (cGxP), гарантируя точное соответствие продукта указанным количествам ингредиентов и маркировке, чтобы все заявления были обоснованными и не вводящими в заблуждение.
В Европейском Союзе регулированием занимается Европейское агентство по лекарственным средствам (EMA) в части централизованного утверждения, в то время как национальные компетентные органы (такие как немецкое BfArM, французское ANSM или CBG/MEB в Нидерландах) занимаются децентрализованными процедурами или процедурами взаимного признания. Для большинства инновационных лекарственных препаратов, особенно тех, которые связаны с биотехнологиями или затрагивают несколько государств-членов ЕС, централизованная процедура является обязательной. Она включает в себя подачу заявки на регистрацию лекарственного средства (MAA), которая должна содержать исчерпывающие данные, аналогичные тем, которые требует FDA, включая клинические и доклинические результаты. Заявления о безопасности и эффективности тщательно изучаются Комитетом EMA по лекарственным средствам для человека (CHMP) до вынесения заключения и предоставления разрешения Европейской комиссией.
В обеих юрисдикциях постмаркетинговый надзор имеет важное значение. Компании обязаны проводить мероприятия по фармаконадзору и могут быть привлечены к проведению дополнительных исследований (испытания IV фазы) для мониторинга долгосрочной безопасности и эффективности. Любые рекламные материалы или объявления должны строго соответствовать утвержденной маркировке и заявлениям; необоснованные или вводящие в заблуждение заявления могут привести к мерам регулирующего воздействия. Кроме того, существуют строгие требования к прозрачности, включая публичное раскрытие данных клинических испытаний.
Хотя системы США и ЕС имеют различные нормативные структуры и процессы, существует значительная гармонизация благодаря международным инициативам, таким как Международный совет по гармонизации технических требований к лекарственным препаратам для человека (ICH) и Соглашения о взаимном признании (подробнее об этом в данном выступлении).
В Нидерландах регулятором фармацевтической отрасли является Совет по оценке лекарственных средств (CBG/MEB). CBG отвечает за оценку и мониторинг безопасности, эффективности и качества лекарственных средств для человека и животных в Нидерландах. Он рассматривает заявки на регистрацию новых лекарственных препаратов и обеспечивает точность, доказательную базу и соответствие нормативной базы информации о продукте и заявленных свойствах. Агентство также участвует в европейской сети регулирования, тесно сотрудничая с Европейским агентством по лекарственным средствам (EMA) и другими национальными органами власти в ЕС.
В обязанности CBG входят:
Выдача разрешений на продажу лекарственных препаратов на национальном уровне в Нидерландах.
Участие в децентрализованных процедурах взаимного признания разрешений на продажу лекарственных препаратов в рамках ЕС.
Участие в централизованных процедурах EMA, включая участие в научных оценках через экспертов в таких комитетах, как CHMP.
Мониторинг фармаконадзора и обеспечение безопасности и эффективности лекарственных средств после их утверждения.
CBG действует под эгидой Министерства здравоохранения, социального обеспечения и спорта Нидерландов и сотрудничает с Инспекцией здравоохранения и по делам молодежи (IGJ), которая следит за соблюдением фармацевтического законодательства и проводит проверки производителей и медицинских учреждений.
2.2. Соглашения о взаимном признании между Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) и Европейским агентством по лекарственным средствам (EMA):
Действия Управления по контролю за продуктами и лекарствами США (FDA) напрямую связаны с действиями Европейского агентства по лекарственным средствам (EMA) в связи с соглашениями о взаимном признании, которые национальные регулирующие органы в сфере пищевых продуктов и лекарств подписали в годы, предшествовавшие 2020 году. Эти соглашения представляют собой контракты, позволяющие одному регулирующему органу принимать без дополнительной проверки то, что другой регулирующий орган в другой стране или регионе предположительно изучил и одобрил. Эта правовая база была создана для содействия гармонизации и эффективности. Однако во время спланированной глобальной пандемии COVID-19 эта структура была использована злоумышленниками в злонамеренных целях, создав ложный фасад регулирования со стороны FDA и позволив другим регулирующим органам просто принимать заявления FDA без проведения обязательной независимой проверки данных.
Вот один из примеров соглашения о взаимном признании (MRA) между FDA и EMA. [8] Это соглашение между США и ЕС вступило в силу 1 ноября 2017 года для лекарственных препаратов для человека и 30 мая 2023 года для ветеринарных препаратов. Оно стало полностью действовать для лекарственных препаратов для человека с 11 июля 2019 года:
«Квалифицированным лицам в государствах-членах ЕС не требуется проводить серийное тестирование лекарственных препаратов для человека, подпадающих под действие Соглашения о взаимном признании лекарственных средств, при условии, что они подтвердили проведение такого контроля в Соединенных Штатах для продукции, произведенной в Соединенных Штатах и импортированной из Соединенных Штатов».
Большинство, если не все, другие национальные системы регулирования пищевых продуктов и лекарственных средств в настоящее время полагаются на правила FDA и их мониторинг соответствия/надлежащей производственной практики (cGMP), не проводя собственных проверок, регулирования, тестирования партий или других процедур cGMP.
2.3. Инъекции мРНК COVID-19 применялись в качестве медицинских контрмер, при этом ложно рекламировались населению как обычно регулируемые фармацевтические продукты.
Хотя инъекции мРНК-вакцин против COVID-19 широко рекламировались как «безопасные и эффективные вакцины», все продукты и протоколы против COVID-19 попали на мировые рынки без соблюдения обычных стандартов фармацевтического производства и распространения, применяемых к регулируемым лекарственным препаратам, из-за их правового статуса «мер противодействия в условиях чрезвычайной ситуации в области общественного здравоохранения». Другими словами, они считаются немедицинскими средствами. Этот статус регулируется совершенно иной, милитаризированной системой управления и отдельным сводом законов, первоначально разработанных для реагирования на атаки с применением химического, биологического, радиологического и ядерного (ХБРЯ) оружия, привязанные ко времени и месту. Ответчики знали об этом. Общественность не была проинформирована о реальном правовом статусе этих инъекций или об их разрешенном использовании.
Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) дает следующее определение медицинских контрмер:
«Медицинские контрмеры (МКМ) — это регулируемые FDA продукты, такие как биологические препараты, лекарства и медицинские изделия, которые могут использоваться в чрезвычайных ситуациях в области общественного здравоохранения для диагностики, профилактики или лечения заболеваний или состояний, вызванных угрозами ХБРЯ или возникающими инфекционными заболеваниями». [9]
Хотя на веб-сайте FDA утверждается, что MCM «регулируются FDA», это утверждение вводит в заблуждение, поскольку в нем упускается тот факт, что MCM не регулируются так же, как обычные фармацевтические продукты, как того ожидают потребители и медицинские работники.
В соответствии с федеральным законодательством США и законодательством ЕС, для медицинского применения лекарственных препаратов FDA/EMA должны официально одобрить любой новый исследуемый препарат, прежде чем производитель сможет ввести его в межштатную торговлю.
Этот процесс требует от производителя подачи заявки на регистрацию нового исследуемого препарата и получения одобрения от FDA/EMA на его использование в регулируемых клинических исследованиях (испытаниях). Поэтому этот регулируемый процесс называется «исследовательским» регуляторным путем. Он обязывает производителя проводить регулируемые клинические исследования, получать одобрение Институционального наблюдательного совета (IRB) для протоколов клинических исследований, обеспечивать независимый мониторинг безопасности и получать информированное согласие от добровольцев, участвующих в клинических исследованиях. Кроме того, производство лекарственных препаратов и биологических препаратов, находящихся в стадии исследования, должно строго соответствовать действующим правилам надлежащей производственной практики (cGMP) и более широким правилам cGxP.
В число противодействий могут входить как ранее одобренные FDA/EMA медицинские изделия (например, опиоиды, аппараты ИВЛ, антибиотики, маски, тампоны и т. д.), так и новые неодобренные продукты (например, инъекции мРНК/ДНК). Однако ключевое различие между противодействиями и обычно регулируемыми лекарственными средствами заключается в законодательной классификации противодействий как не подлежащих клиническим исследованиям . В частности, в законодательстве США использование противодействий, разрешенных к экстренному применению (EUA), в условиях объявленной чрезвычайной ситуации в области общественного здравоохранения не может считаться клиническим исследованием (21 USC 360bbb-3(k)), поэтому противодействия не могут быть протестированы на безопасность или эффективность в соответствии с законодательством США (21 CFR 312 и 21 CFR 601), и FDA не может обеспечить соблюдение действующих правил надлежащей производственной практики (cGMP) или надлежащей практики дистрибуции (GxP в целом).
Эта переклассификация ранее одобренных и неодобренных медицинских изделий позволяет использовать средства противодействия токсичным веществам (СППВ) для «неодобренных целей». Вместе со статусом «не находящееся на стадии исследования» эта переклассификация исключает использование СППВ в качестве лекарственных средств и, устраняя меры защиты потребителей, делает СППВ полностью уязвимыми для использования в качестве оружия.
Законы, позволяющие отменить все меры защиты потребителей и ответственность производителей, включают Закон о PREP (2005 г.) [10] , статью 564 Закона о пищевых продуктах, лекарствах и косметике (FDCA) [11] в США и ряд положений ЕС о «готовности к пандемии» и «медицинских мерах противодействия» (обсуждаются в разделе 2.5). Закон о PREP и соответствующие положения ЕС о мерах противодействия отменяют применение фармацевтического законодательства к продуктам, объявленным мерами противодействия в рамках существующего чрезвычайного положения, и предоставляют иммунитет от ответственности лицам, подпадающим под действие закона (за исключением узко определенных умышленных противоправных действий). Закон о PREP является предметом юридических споров, и в настоящее время в Конгрессе США рассматривается предложение о его отмене [12] .
Ввиду заявленного неисследовательского статуса медицинских средств противодействия, хотя производители могут по своему усмотрению, а FDA/EMA могут потребовать выполнения некоторых действий, обычно ожидаемых от исследовательских клинических испытаний и процесса валидации производства, ни один из типичных фармацевтических нормативных стандартов не применим в принудительном порядке. В целом, любые действия, заявленные как регулируемые исследовательские испытания и процессы для медицинских средств противодействия, следует рассматривать как обманные действия, направленные на создание видимости защиты потребителей там, где ее нет и не предполагается.
FDA имеет право выдавать разрешение на экстренное использование, если, по единоличному мнению министра здравоохранения и социальных служб, продукт «может быть эффективным» в лечении соответствующего заболевания или состояния [13] . Никакие другие критерии одобрения не применяются в принудительном порядке. Нет строгих юридических требований к проведению клинических испытаний до выдачи разрешения, и проведение законных клинических испытаний с участием людей невозможно из-за установленного законом «неисследовательского» статуса контрмер.
FDA одобрит меры противодействия EUA на основании неполной/отсутствующей информации, исходя из мнения министра здравоохранения и социальных служб о том, что «известная и потенциальная польза продукта» может «перевешивать известные и потенциальные риски» [14] и считает маловероятным наличие «полных данных об эффективности» до выдачи разрешения на EUA. В отличие от этого, в отношении исследуемого препарата (в рамках обычного процесса регулирования) FDA «должно» отказать в одобрении, если заявитель «не докажет, что такой препарат безопасен» [15] .
Строгих требований к получению разрешения на проведение клинических исследований нового лекарственного препарата (IND) нет, как и к одобрению протокола клинического исследования и форм информированного согласия этическим комитетом (IRB). Следовательно, статус экстренного разрешения (EUA) для медицинского средства защиты исключает сбор данных регулируемых клинических исследований и, таким образом, исключает получение надежных и достоверных научных знаний о рисках и преимуществах, связанных с этим средством защиты, пока оно остается не на стадии исследования.
Кроме того, отсутствуют обязательные стандарты контроля качества в производстве; нет проверок производственных процедур; нет испытаний при выпуске партий и нет запрета на значительную вариативность между партиями; нет запрета на фальсификацию; и нет обязательного соблюдения действующих правил надлежащей производственной практики. Продукция, разрешенная к экстренному использованию, даже несмотря на то, что она не регулируется и не стандартизирована, «не должна считаться фальсифицированной или неправильно маркированной». [16]
Путь разрешения на экстренное использование медицинских контрмер применяется только тогда, когда министр здравоохранения и социальных служб США или министр здравоохранения государства-члена ЕС объявляет чрезвычайное положение [17] . В Соединенных Штатах действие чрезвычайных положений Закона PREP в связи с COVID-19 продлевалось 12 раз и действует до конца 2029 года [18] , что предотвращает введение обязательных к исполнению правил и расширяет защиту от ответственности за смерть и травмы, вызванные контрмерами.
В Нидерландах правительство официально классифицировало COVID-19 как инфекционное заболевание категории А в соответствии с национальным Законом об общественном здравоохранении (Wet publieke gezondheid) 27 января 2020 года. Эта классификация, объявленная министром здравоохранения Бруно Брюинсом, активировала ряд чрезвычайных мер. Этот шаг фактически представляет собой национальное объявление чрезвычайной ситуации в области общественного здравоохранения, наделяющее власти полномочиями в соответствии с Законом об общественном здравоохранении. Эти полномочия позволили принять общенациональные меры реагирования, такие как локдауны, без объявления официального чрезвычайного положения. Нидерланды никогда не объявляли официальное национальное «чрезвычайное положение в области общественного здравоохранения»; следовательно, нет никакого чрезвычайного положения, которое нужно было бы официально отменить.
Вкратце, как в США, так и в ЕС, правовой статус продукта, услуги, процедуры или действия, обозначенных как «контрмера», эквивалентен статусу потенциального оружия. Лекарственные препараты или потенциальные лекарственные препараты (неразрешенные лекарства), обозначенные как «контрмеры» в условиях реальной или сфабрикованной чрезвычайной ситуации, имеют законное право использоваться в немедицинских целях. К немедицинским целям относится использование в качестве оружия или незаконный эксперимент над людьми. В соответствии с действующим законодательством допускается искажение информации о безопасности, эффективности или составе продуктов, разрешенных к применению в рамках чрезвычайного положения. Ответчики знали или должны были знать об этом, но скрыли это от общественности и распространяли чудовищную ложь и принуждение, чтобы добиться максимального распространения этих мнимых лекарств, которые на самом деле являются биохимическим оружием.
2.4. Правовые положения о контрмерах в ЕС.
Хотя соответствующие законы США и ЕС не идентичны, существует ряд положений ЕС, соответствующих положениям США в отношении «медицинских контрмер».
В Европейском союзе (ЕС) медицинские контрмеры регулируются в основном в рамках законодательства ЕС в области фармацевтики, медицинских изделий и общественного здравоохранения, особенно в части готовности к серьезным трансграничным угрозам здоровью. Хотя ЕС не использует термин «медицинская контрмера» так явно, как это делает Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA), правовая инфраструктура поддерживает разработку, авторизацию, создание запасов и развертывание таких продуктов в чрезвычайных ситуациях в области общественного здравоохранения.
Основные законы и регламенты ЕС, регулирующие медицинские контрмеры:
1. Регламент (ЕС) 2022/2371 «О серьезных трансграничных угрозах здоровью»
Принято: 2022 год
Устанавливает:
План готовности к кризисам и пандемиям в сфере здравоохранения
Координация медицинских контрмер на уровне ЕС
Управление по обеспечению готовности и реагирования на чрезвычайные ситуации в области здравоохранения (HERA)
2. Регламент (ЕС) 2022/123 «Об усилении роли Европейского агентства по лекарственным средствам (EMA)»
Принято: январь 2022 года
Цель: Расширяет роль EMA в мониторинге, координации и содействии разработке и доступности лекарственных средств и медицинских изделий во время чрезвычайных ситуаций в области общественного здравоохранения.
Создает Руководящую группу по дефициту лекарственных средств (MSSG) и Чрезвычайную целевую группу (ETF).
Правовая основа для выдачи разрешений на экстренное использование средств противоракетной обороны в ЕС.
Правовая справка: Регламент (ЕС) 2022/123
3. Соглашение о совместных закупках (СПЗ) в соответствии со статьей 5 Решения 1082/2013/ЕС
Позволяет государствам-членам ЕС совместно закупать вакцины , противовирусные препараты , средства индивидуальной защиты и другие средства противодействия чрезвычайным ситуациям.
Инструмент скоординированных закупок продолжает функционировать и после пандемии COVID-19 в рамках программы HERA.
4. В 2016 году ЕС принял Регламент (ЕС) 2016/369, https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32016R0369&from=EN , который предусматривает экстренную поддержку в рамках Европейского союза. Экстренная поддержка «может предоставляться посредством специальных мер, соответствующих экономической ситуации, в случае продолжающейся или потенциальной природной или техногенной катастрофы » (ст. 1 (1)). На основании решения Совета в соответствии со ст. 2 Регламента 2016/369/ЕС « активировать экстренную поддержку » в рамках этого Регламента, после предложения, представленного Комиссией ЕС, были созданы условия для финансирования « специальных мер » против угрозы, возникающей в результате « техногенных или природных катастроф ».
14 апреля 2020 года в настоящее Положение были внесены поправки Регламентом (ЕС) 2020/521 [19] с обратной силой с 1 февраля 2020 года , чтобы расширить применение настоящего Регламента на оказание экстренной помощи во время кризиса COVID-19.
В статье 1 активируется « чрезвычайная поддержка в соответствии с Регламентом Совета (ЕС) 2016/369 ».
Настоящий Регламент предусматривает приложение к Регламенту 2016/369/ЕС, в котором перечисляются допустимые меры, которые могут быть профинансированы в случае чрезвычайных ситуаций. В нем указывается, что этот перечень не является исчерпывающим, и он гласит:
« В случае пандемий, имеющих масштабные последствия, могут быть профинансированы следующие меры : »
(а) временное усиление медицинского персонала, обмен медицинскими специалистами, прием иностранных пациентов или другие виды взаимной поддержки;
(б) развертывание временных медицинских учреждений и временное расширение существующих медицинских учреждений для снижения нагрузки на существующие структуры и увеличения общей пропускной способности системы здравоохранения;
(c) деятельность по поддержке администрирования широкомасштабного применения медицинских тестов и подготовке необходимых научных стратегий и протоколов тестирования;
(d) создание временных карантинных учреждений и принятие других соответствующих мер на границах Союза ;
(e) разработка, производство, закупка и распространение медицинских изделий;
(f) увеличение и перепрофилирование производственных мощностей по выпуску медицинской продукции, как указано в пункте (e), для решения проблемы нехватки поставок;
(g) поддержание запасов медицинских изделий, указанных в пункте (e), и их утилизация;
(h) действия по поддержке необходимых шагов для получения разрешения на использование медицинских изделий, указанных в пункте
(e), если это требуется;
(i) действия по разработке соответствующих методов отслеживания развития пандемии и результатов мер,
принятых для борьбы с ней;
(j) организация внеплановых клинических испытаний потенциальных методов лечения или диагностики в соответствии со стандартами испытаний, согласованными на уровне Союза;
(k) научная проверка медицинских изделий, включая потенциальные новые методы тестирования.
Приведенный выше список не является исчерпывающим.
Таким образом, (h) позволяет предпринять любые действия, необходимые для получения разрешения на использование медицинских изделий. Это позволяет избежать применения любых положений, применимых к лекарственным препаратам и медицинским изделиям.
В пункте (j) показано, что для «специальных» клинических испытаний законы о надлежащей клинической/надлежащей производственной практике (GCP) могут быть отменены и заменены довольно расплывчато определенными «стандартами испытаний, согласованными на уровне Союза», то есть правилами, согласованными неизбранными бюрократами, а не фармацевтическими нормами, изложенными в законе, регулирующем безопасность потребителей медицинских изделий.
2.5. В ЕС для принятия контрмер под видом лекарственных средств использовался механизм условного разрешения на продажу (Conditional Marketing Authorization, CMA).
Обширные доказательства обмана общественности со стороны фармацевтических компаний, действующих в сговоре с регулирующими органами, стали доступны в конце ноября 2020 года, когда из EMA [20] просочилось около 1000 страниц документации по контролю качества химического производства (CMC) и ряд внутренних электронных писем . Полный набор просочившихся страниц и писем включен в Приложение к этому заявлению. Эти документы продемонстрировали, что должностные лица здравоохранения Европейского союза совершили мошенничество в отношении всех граждан, создавая впечатление, что они оценивали и одобряли инъекции от COVID-19 в соответствии с существующими стандартами для лекарственных средств, по крайней мере, на основе условных разрешений на маркетинг (CMA). Документы показали, что за кулисами регулирующие органы были озабочены исключительно сроками запуска, еще до того, как были рассмотрены какие-либо данные, и отменяли или значительно снижали стандарты качества, чтобы помочь Pfizer «соответствовать» этим стандартам, основываясь на заявлении Pfizer и без какой-либо научной основы. Эта обманная схема «подмены товара» координировалась на глобальном уровне между регулирующими органами – FDA, EMA, MHRA, TGA, Health Canada и многими другими.
В ЕС тот же самый конечный эффект — отсутствие какого-либо подлежащего исполнению соблюдения фармацевтических правил cGxP и других соответствующих фармацевтических законов — был достигнут путем принуждения всех государств-членов к подписанию организованных ЕС хищнических контрактов на закупку с фармацевтическими производителями, которые отменяли все соответствующие фармацевтические правила и обязывали национальные правительства возмещать производителям убытки в случае успешных исков об ответственности против фармацевтических компаний в государствах-членах. Это возмещение включало отказ от суверенного иммунитета (см. стр. 32 Соглашения о предварительной закупке Pfizer с ЕС [21] ). Эти контракты фактически препятствовали государствам-членам осуществлять суверенные законодательные полномочия в отношении фармацевтической ответственности за эти продукты в своих странах. Эти контракты якобы были заключены Урсулой фон ден Лейен посредством текстовых сообщений. В настоящее время Европейская прокуратура (ЕППО) проводит расследование по поводу приобретения вакцин от COVID-19 в Европейском союзе [22] . Ответчики, выступавшие в качестве соответствующих органов здравоохранения в Нидерландах, знали или должны были знать, что эти договорные положения носили хищнический характер, нарушали конституцию соответствующего государства и, следовательно, были неприемлемы для суверенного правительства, а также привели бы к массовым травмам среди граждан, которых эти должностные лица органов здравоохранения поклялись защищать.
Одним из важнейших механизмов обмана было привязывание всех 27 государств-членов ЕС к одной (слепой) сделке путем обещания, что вакцины в Европе пройдут через процедуру условного разрешения на продажу (CMA), а не через экстренное разрешение, то есть вопрос статьи 5(2) против CMA. Это обсуждается в нескольких электронных письмах, включенных в утечку документации EMA. Например:

Статья 5(2) представляет собой механизм экстренного разрешения, доступный отдельным государствам-членам. Это разрешение выдается только на один год, выдается каждым государством, и каждое государство может отозвать его самостоятельно. Механизм CMA является общеевропейским механизмом, в котором отдельные государства не имеют права голоса. Однако в ЕС механизм CMA представляет собой неэкстренный исследовательский путь, который до 2020 года использовался только для онкологических препаратов в рамках «права на попытку» или «гуманного использования» для неизлечимо больных пациентов. Он аналогичен механизму расширенного доступа к лекарственным средствам [23] (EAU, не путать с EUA) в США. Однако оба пути, CMA и EAU, являются исследовательскими, то есть юридически обозначены как лекарственное средство и предназначены для медицинского применения. Однако, как обсуждалось выше, «контрмеры» — это неисследовательские вещества, предназначенные для немедицинских целей, поэтому обозначение их как CMA или «BLA» является мошенничеством и обманом, связанным с их использованием в качестве оружия.
Как указано в законе, CMA звучит как гораздо более строгий механизм регулирования и соблюдения требований по сравнению со статьей 5(2):

Важное различие между статьей 5(2) и CMA заключается в том, что она возлагает ответственность на производителя, и именно это было обещано обеспечить посредством общеевропейского разрешения CMA:

Однако в классической мошеннической схеме обмана и подмены товаров органы здравоохранения ЕС и Нидерландов никогда не намеревались обязывать фармацевтические компании соблюдать обещанные стандарты CMA. Контракты ЕС на поставку фармацевтической продукции предусматривали отмену всех соответствующих правил и законов по обеспечению безопасности потребителей в этих странах. Фактически это лишило стандарты CMA смысла в отношении безопасности потребителей и ответственности производителей, поскольку отсутствие контроля означает отсутствие закона!
Например, см. соглашение ЕС о поставках с Pfizer [24] в отношении возмещения убытков:
В итоге, чистый эффект Закона о профилактике и профилактике заболеваний (т.е. отмена всех соответствующих фармацевтических правил и ответственности производителей) был достигнут в ЕС путем централизации соглашений о поставках в рамках общеевропейской мошеннической схемы, разработанной для перекладывания военных контрмер на потребителей под видом вакцин, одобренных CMA, а затем и «полностью утвержденных».
2.6. Прямые доказательства несоответствия инъекций мРНК/ДНК COVID-19 обычным фармацевтическим нормам, ожидаемым потребителями и медицинскими работниками.
Примерно 1000 страниц производственной документации Pfizer были слиты из Европейского агентства по лекарственным средствам (EMA) в конце 2020 года, что свидетельствует об отсутствии соответствия надлежащей производственной практике (cGMP) менее чем за две недели до начала поставок продукта миллиардам потребителей по всему миру. Подлинность утечки документов была подтверждена Британским медицинским журналом . EMA не отрицало подлинность документов. Помимо производственной документации, файлы EMA также содержат 14 скриншотов электронных писем, датированных серединой-концом ноября 2020 года. Переписка по электронной почте охватывает сотрудников EMA и высшее руководство агентства, а также их переписку с FDA и MHRA (британским регулятором). Эти электронные письма демонстрируют, что эксперты EMA находились под огромным политическим давлением с целью изобретения новых способов одобрения заведомо опасных продуктов, которые не подлежат одобрению. Из электронных писем, комментариев и возражений, высказанных сотрудниками EMA, очевидно, что они не были осведомлены о правовом статусе инъекций COVID-19 в качестве средств противодействия, а также о том, что регуляторная проверка данных не имела существенного значения для их применения.
Также очевидно, что руководство EMA было в первую очередь озабочено координацией дат запуска, и «разрешение» на эти испытания для всех государств-членов ЕС было предрешено. Давление с целью игнорирования всех недостатков в регулировании исходило с самого верха правительств США, Великобритании и ЕС.
Ввиду намеренно нереалистичных сроков, существовали серьезные и неразрешимые проблемы с качеством продукции, которую сотрудники EMA были вынуждены одобрить. Производственный процесс был вопиюще не соответствовал требованиям. Эксперты EMA выдвинули более 100 возражений против одобрения, включая несколько серьезных возражений. В частности, к серьезным возражениям относились:
1) несоответствие требованиям надлежащей производственной практики (cGMP);
2) отсутствие целостности мРНК и большое количество фрагментов мРНК ( некоторые из которых можно охарактеризовать как miRNA, siRNA и shRNA — все это потенциальные компоненты для создания оружия, как описано выше );
3) многочисленные существенные пробелы в производственной документации, делающие невозможным определить, можно ли изготовить продукт в соответствии с описанием.
Утечка электронных писем из EMA также демонстрирует, что регуляторы ЕС были озабочены только датами запуска продукта и не собирались проверять необходимые данные до запуска. Процесс был сугубо политизированным, а не научным. Регуляторы EMA получили устные заверения от должностных лиц FDA вместо того, чтобы проверить данные. Как указано выше, сотрудники EMA, занимавшиеся проверкой данных, возражали против них, но высокопоставленные чиновники проигнорировали и отклонили их возражения.
Отсутствие соответствия стандартам cGMP означает, что невозможно гарантировать наличие в отправляемых пациентам препаратах определенных ингредиентов в определенных количествах. Следовательно, помимо опасности препарата, получение информированного согласия невозможно.
Одним из ключевых элементов, гарантирующих качество лекарственного вещества, является спецификация продукта, основанная на текущем состоянии разработки продукта, а также науке и технике. Для этого установлены рамки руководства Международной конференции по гармонизации (ICH) Q6A «Процедуры испытаний и критерии приемлемости для новых лекарственных веществ и новых лекарственных препаратов: химические вещества» [25] . Спецификация определяет перечень испытаний, а также ссылки на аналитические процедуры и соответствующие критерии приемлемости для оценки качества соответствующего продукта в отношении идентичности, содержания/количества, чистоты/примесей и активности/биологической активности.
Все аналитические процедуры должны быть полностью валидированы, что означает использование либо стандартных, ранее валидированных методов, либо, если это запатентованные/новые методы, то производитель должен разработать и полностью валидировать анализы, используемые для новых методов. Это требует, помимо прочего, проведения точных тестов с положительными и отрицательными контролями, а также полной и отслеживаемой документации («аудиторского следа») для каждого теста. Коммерческая тайна не является оправданием для несоблюдения этих требований. Даже если производитель не желает публично раскрывать свои аналитические процедуры, он все равно должен предоставить полную прозрачную документацию в FDA, которое хранит ее в архиве (и этот факт официально сообщается покупателям продукта для обеспечения их соответствия требованиям GxP).
В документации Pfizer по контролю качества и производству, просочившейся в конце 2020 года, эксперты EMA отметили отсутствие описания «некомпендиальных», то есть собственных запатентованных методов аналитических процедур Pfizer. Это сделало невозможным оценку научной точности запатентованных методов, используемых Pfizer для контроля качества производимых инъекционных препаратов.
Многочисленные параметры контроля продукции и процессов были запатентованы, недостаточно четко определены, некоторые еще не были изобретены, и ни один из них не был научно подтвержден. Например, для определения идентичности мРНК был выбран метод ОТ-ПЦР (ПЦР в реальном времени). Однако мРНК — это крайне нестабильное и хрупкое вещество, которое, несмотря на манипуляции и химическую «оптимизацию» для обеспечения стабильности, все же демонстрирует высокую степень хрупкости. В результате целостность мРНК оказалась проблематичной, особенно при масштабировании производства.
Одно из главных замечаний экспертов EMA, изложенных в просочившейся в СМИ документации Pfizer по контролю качества и производству, касалось недостаточной целостности мРНК, то есть низкого процента РНК во флаконе, соответствующей спецификации, и очень высокого процента поврежденных фрагментов РНК.

Большое количество несоответствующей РНК было признано экспертами EMA примесью. Это действительно серьезная проблема — вся заявленная эффективность продукта якобы зависела от «правильного кода» для производства «уханьского шиповидного белка» с использованием клеточного механизма инъецируемых лиц. Здесь мы видим доказательства того, что регулирующие органы возражают против одобрения из-за того, что в клетках, подвергнутых воздействию фрагментов РНК, могут происходить многие другие процессы, помимо образования «уханьского шиповидного белка», что является известным методом создания мРНК-вакцин, как обсуждалось выше .
Вместо того чтобы приостановить утверждение и потребовать устранения проблемы с целостностью мРНК, регулирующие органы просто произвольно повысили критерий приемлемости для процента целостности мРНК с предыдущего стандарта 70%+ до чуть более 50%.
Это означает, что значительная часть лекарственного вещества может содержать «мусорный» РНК-материал, фрагменты и нехарактеризованные фрагменты, некоторые из которых достаточно велики, чтобы кодировать неизвестные и, возможно, аберрантные белки, и большинство из них относятся к категории микроРНК (миРНК). Эти короткие последовательности, хотя и не кодируют, как известно, вмешиваются в клеточные процессы и участвуют в онкологических процессах [26] .
В конечном итоге, сама проверка регулирующих органов и возражения, высказанные экспертами EMA, не имели значения — продукт все равно был бы выпущен на рынок, несмотря на возражения регулирующих органов, из-за применения военной химического, биологического, радиологического и ядерного оружия в глобальном масштабе, втайне от общественности.
2.7. Отмена требований к проверкам предприятий по производству биопрепаратов:
Ни FDA, ни EMA не проводили инспекции производственных мощностей Pfizer и ее поставщиков в 2020 году, ссылаясь на чрезвычайную ситуацию, связанную с COVID-19. Когда инспекции производственных мощностей возобновились в 2022 году, было обнаружено, что основной европейский подрядчик Pfizer, компания Rentschler, нарушила правила надлежащей производственной практики (выдана форма 483). Это означает, что цепочка поставок Pfizer в Европе не соответствовала требованиям в период с 2020 по 2022 год. Регуляторы не предприняли никаких мер принудительного характера, поскольку, согласно закону об экстренном разрешении на использование, принудительное исполнение невозможно.
Также стоит отметить, что в 2019 году, очевидно, в рамках подготовки к операции по борьбе с COVID-19, тогдашний комиссар FDA США Скотт Готтлиб изменил федеральные правила, регулирующие инспекцию лицензированных предприятий, производящих все биологические продукты, включая «вакцины» , с периодичности инспекций не реже одного раза в два года на неопределенный срок; отменил положения о принудительном исполнении в случае, если лицензированное предприятие не прошло инспекцию; и отменил все обязанности инспекторов FDA по проведению инспекций. До изменения правил, пункт 21 CFR 600.21, «Время инспекции», гласил:
«Проверка предприятия, на которое подана заявка на получение лицензии на производство биологических препаратов, может быть проведена только после того, как предприятие начнет работу и будет производить полный ассортимент продукции, для которой запрашивается лицензия на производство биологических препаратов».
В случае отказа в выдаче лицензии после проверки первоначальной лицензии, повторная проверка не требуется до получения подтверждения об устранении выявленных недостатков, послуживших основанием для отказа. Проверка каждого лицензированного заведения и его дополнительных объектов должна проводиться не реже одного раза в 2 года. Проверки могут проводиться с предварительным уведомлением или без него и должны проводиться в обычные рабочие часы, если не указано иное.
С 2 мая 2019 года последние три предложения раздела 21 CFR 600.21 были удалены.
В настоящее время нет законодательного требования о проведении первоначальной инспекции FDA; нет минимального интервала для последующих инспекций FDA, и нет никаких юридических последствий за несоблюдение требований, таких как отказ в выдаче или аннулирование лицензии на предприятие или продукт.
Правовые механизмы, посредством которых исчезло регулирование FDA производства биологических продуктов, включали в себя одновременно опубликованные в Федеральном реестре 26 февраля 2018 года окончательное правило и проект правила, а также окончательное правило от 2 апреля 2019 года, изданное тогдашним комиссаром FDA Скоттом Готтлибом.
2.8. Произвольная переклассификация генной терапии на основе мРНК/ДНК как «вакцин от COVID-19»:
Что касается самой технологической платформы (мРНК в липидных наночастицах [ЛНП] или ДНК в аденовирусном векторе) — обе известны как технологии «трансфекции». Цель разработки продукта — доставить различные биохимические вещества внутрь клеточных мембран, а часто и в ядро клетки, где находится ДНК. Методы трансфекции с использованием широкого спектра РНК и ДНК-технологий являются хорошо зарекомендовавшей себя научной реальностью. Например, как опубликовано в этой обзорной статье [27] :
« Для выявления соответствующих опубликованных исследований или протоколов, подпадающих под рамки данного обзора, был проведен систематический поиск литературы на основе рекомендаций PRISMA ( Moher et al., 2009 ) ( рис. 1 ). Для поиска литературы использовались базы данных Scopus, Google Scholar и PubMed. Ключевые слова, использованные в ходе поиска, включали «трансфекция», «котрансфекция», «химические вещества», «реагенты», «ДНК», «siRNA», «shRNA», «miRNA», «плазмида», «олигонуклеотиды» , «эффективность», «безопасность», «цитотоксичность», «контроли» и другие связанные ключевые термины. Первоначальный поиск выявил около 5000 статей, опубликованных протоколов или руководств из различных баз данных, содержащих описания или сравнения различных методов трансфекции, типов трансфицированных нуклеиновых кислот, контроля трансфекции, методов оценки эффективности трансфекции и реагентов для трансфекции ».
Конструкция продуктов генной терапии идентична конструкции «вакцин» от COVID-19: липосомальные наночастицы (ЛНП) будут доставлять прикрепленный к ним груз в клетки, то есть трансфицировать клетки . Платформа ЛНП или платформа аденовирусов — это всего лишь два разных типа «грузовиков», причем ЛНП особенно эффективны для взлома клеток. Тот факт, что это технология трансфекции, широко описан в научной литературе и в нормативных документах. Компании Moderna и BioNTech в документах, поданных в Комиссию по ценным бумагам и биржам США (SEC) непосредственно перед пандемией, характеризовали свои мРНК-технологии как экспериментальные, «терапевтические» и находящиеся на ранних стадиях разработки. В отчете Moderna за 2019 год по форме 10-K конкретно отмечалось: «мРНК-препараты представляют собой новый и непроверенный подход... Ни одна мРНК-иммунотерапия не была одобрена, и ни одна, возможно, никогда не будет одобрена».
До 2020 года ни один фармацевтический препарат на основе мРНК не был одобрен ни одним регулирующим органом в мире. Это было связано с многочисленными нарушениями стандартов безопасности и соответствия фармацевтическим стандартам. Все разрабатываемые продукты неоднократно сталкивались с проблемами безопасности и не могли продвинуться даже до первой фазы клинических испытаний на людях [28] , [29] .
3. Инъекции мРНК COVID-19 финансировались, разрабатывались и применялись по всему миру в рамках военной кампании, координируемой военными и силовыми альянсами.
3.1. COVID-19 был не событием, связанным с общественным здравоохранением, а глобальной военной операцией:
Операция по борьбе с COVID-19 не была событием, связанным с общественным здравоохранением, хотя и представлялась таковой населению мира. Это была глобальная операция, координируемая посредством государственно-частных разведывательных и военных альянсов и включающая в себя законы, разработанные для атак с применением химического, биологического, радиологического и ядерного оружия (ХБРЯ).
В США за политику в отношении COVID-19 отвечает Совет национальной безопасности (а не Министерство здравоохранения и социальных служб). В других странах мира кампания по борьбе с COVID-19 координировалась с помощью идентичных механизмов: военные и разведывательные службы руководили политикой реагирования (как во время войны), а органы здравоохранения создавали ложное впечатление о чрезвычайной ситуации в области общественного здравоохранения. См. Приложение 1 для получения подробных доказательств глобальной военной кампании, включая данные по Нидерландам и другим странам ЕС.
В США, 13 марта 2020 г.: «Адаптированный план реагирования правительства США на COVID-19 PanCAP» (PanCAP-A) гласит, что политика Соединенных Штатов в ответ на SARS-CoV-2 определяется не органами здравоохранения, указанными в протоколах готовности к пандемии (Закон о готовности к пандемиям и всем видам чрезвычайных ситуаций, [30] PPD-44, [31] BIA), а Советом национальной безопасности (СНБ). СНБ не принимает постоянных представителей органов здравоохранения, и его деятельность сосредоточена на вопросах национальной безопасности и внешней политики».
Ниже представлена организационная схема из документа PanCAP-A, стр. 9:

3.2. Организационная структура операции «Варп-скорость»
Согласно отчетам Operation Warp Speed/ASPR, Operation Warp Speed была объявлена «совместными» усилиями Министерства обороны и Министерства здравоохранения и социальных служб по созданию «безопасных и эффективных» вакцин и терапевтических средств против COVID-19. Однако, согласно организационной схеме, Министерство обороны формально являлось главным операционным директором, а Министерство здравоохранения и социальных служб занимало должность главного научного советника. [32]

VRBPAC-10.22.20-Meeting-Presentation-COVID19-Vaccine-Development-Portfolio.pdf
Следует отметить, что следующий по старшинству уровень организации контролируется правительством США и включает в себя все надзорные функции в сфере производства, разработки и проведения клинических испытаний, планирования операций и анализа, дистрибуции, связей с общественностью, заключения контрактов, юридических и других функций. Фармацевтические компании находятся на третьем уровне ниже в этой организации.
В отчете STAT News за 2020 год указывалось, что в руководстве операции Warp Speed участвовало около 60 военных чиновников, включая четырех генералов, многие из которых не имели никакого предыдущего опыта работы в сфере здравоохранения. Из примерно 90 руководящих должностей в организационной структуре только 29 не были сотрудниками Министерства обороны. [33]
Рассекреченные документы октября 2020 года, представленные на презентациях Operation Warp Speed в Консультативном комитете FDA по вакцинам и связанным с ними биологическим продуктам, раскрывают контроль правительства США над почти всеми аспектами проектирования и внедрения продуктов для борьбы с COVID-19 [34] .
3.3. Обзор контрактов Министерства обороны США и BARDA на меры по борьбе с COVID-19
Сотни контрактов на меры противодействия COVID-19 стали доступны через запросы «о свободе информации» (FOIA) в частично отредактированном виде. [35] Анализ этих контрактов указывает на высокую степень контроля со стороны правительства США (DoD/BARDA) и определяет объем поставляемых товаров только как «демонстрации» и «прототипы». Демонстрация — это фиктивная, показная деятельность. Лекарственные препараты, применяемые к людям, нельзя характеризовать как «демонстрации и прототипы», однако оружие может быть заказано как прототип. Контракты также включают снятие всей ответственности с производителей и любых подрядчиков по цепочке поставок и распределения в соответствии с Законом PREP 2005 года и соответствующим федеральным законодательством.
Хотя в контрактах Министерства обороны и BARDA, касающихся контрмер, упоминаются требования к безопасности и эффективности вакцин, а также соответствие действующим правилам надлежащей производственной практики (cGMP), эти пункты явно исключены из перечня товаров, которые не оплачиваются (или не заказываются) правительством США.
Контракты были структурированы в рамках процедуры «Другие транзакции» (Other Transactions Authority, OTA) — метода заключения контрактов, который использовался Министерством обороны и BARDA для всех мер противодействия COVID-19, заказанных у частной промышленности. Метод OTA позволяет федеральным агентствам заказывать продукцию, подлежащую регулированию, минуя любые подобные правила, а также механизмы финансовой подотчетности, которые охватывают стандартные государственные контракты и другие законы, регулирующие раскрытие информации и интеллектуальную собственность (ИС), полученную в результате исследований, финансируемых из государственных средств. [36]
«Прочее» — это обобщающая категория, не включающая контракты, исследовательские гранты, закупки и т. д.: это не какие-либо обычно регулируемые/подотчетные государственные контракты.
Министерство обороны использовало OTA для заказа расплывчато определенных «прототипов» и «демонстраций», которые не подлежат контролю со стороны регулирующих органов.
В собственном отчете BARDA продукция, связанная с COVID-19, отнесена к категории «демонстрационных образцов» или, в лучшем случае, «крупномасштабного производства»:

3.4. Контрмеры Министерства обороны/BARDA, разработанные устоявшейся сетью оборонных подрядчиков.
Контракты Министерства обороны/BARDA на «меры противодействия» находятся в ведении Advanced Technology International (ATI). [37] ATI в основном управляет консорциумами НИОКР для Министерства обороны по таким направлениям, как производство оружия, литье и ковка металлов, производство кораблей и технологии, направленные на «противодействие оружию массового уничтожения (ОМУ)». Два из этих консорциумов связаны с биомедицинскими проектами.
Консорциум медицинских технологических предприятий (MTEC), действующий от имени Командования медицинских исследований и разработок армии США, занимается разработкой технологий для редактирования генов, нанотехнологий, «телемедицинских решений», искусственных конечностей и мозговых имплантатов. В настоящее время MTEC разрабатывает носимое устройство для диагностики COVID-19 до появления симптомов.
В состав Медицинского консорциума по защите от химического, биологического, радиологического и ядерного оружия (MCDC) [38] в настоящее время входят более 300 крупных и малых предприятий и академических организаций, которые «поддерживают потребности Министерства обороны (DoD) в медицинской фармацевтике и диагностике для противодействия химическим, биологическим, радиологическим и ядерным угрозам (ХБРЯ)» и обеспечивают «прототипные технологии для терапевтических медицинских контрмер, нацеленных на вирусные, бактериальные и биологические токсины, представляющие интерес для DoD», включая разработку вакцин.
В рамках механизма «Уполномоченных по другим сделкам» MCDC заключил контракты с сотнями компаний на поставку «мер противодействия» COVID-19. Заказ на дозы вакцины Pfizer был размещен 20 июля 2020 года в рамках базового соглашения между Advanced Technologies Inc (ATI, компания по управлению поставщиками Министерства обороны США) и Pfizer, Inc., обозначенного как базовое соглашение MCDC № 2020-532:
· 21 июля 2020 г., Письмо с техническими указаниями MCDC или Техническое задание (SOW) для «Демонстрации крупномасштабного производства вакцины против пандемии COVID-19» между Pfizer и Министерством обороны/Advanced Technologies Inc. [39]
В контрактах было указано, что продукция будет поставляться Министерству обороны в качестве единственного покупателя. Поставляемая продукция не имеет серийных номеров – то есть, на дозированных упаковках нет штрихкодов, и, следовательно, ее невозможно отследить в соответствии с обычными правилами дистрибуции фармацевтической продукции, которые существуют для выявления любых проблем с безопасностью или качеством в цепочке поставок. Таким образом, продукция подвержена как фальсификации/неправильной маркировке, так и подделке. Продукция была отправлена в Министерство обороны и обрабатывалась через «черный ящик» системы дистрибуции Министерства обороны, якобы из-за требований к хранению в условиях холодовой цепи, которые впоследствии были отменены, но практика дистрибуции через военных подрядчиков и по военным контрактам не изменилась.
Продукт был обозначен как «собственность правительства США» [40] до момента его введения человеку. Все лица, выполняющие любые задачи, связанные с производством, цепочкой поставок, распределением и применением вакцин, являются «лицами, подпадающими под действие» Закона PREP, если они выполняют распоряжения правительства США. Независимо от места работы, они считаются сотрудниками правительства США для целей данной работы.
Важно отметить, что в контрактах Министерства обороны меры по борьбе с COVID-19 описываются как предназначенные для «гражданского и военного применения».
4. Выводы и мнение экспертов:
На основании анализа основных нормативных документов, просочившихся в прессу файлов Pfizer по контролю качества химического производства (CMC), соответствующего законодательства США и ЕС, а также другой общедоступной документации, я, как эксперт, пришел к выводу, что инъекции мРНК COVID-19 применялись в соответствии с военными правилами «медицинского противодействия», которые обошли стандартные фармацевтические меры безопасности, что делает их юридически и функционально неотличимыми от потенциального биохимического оружия.
Вакцины от COVID-19 были обманным путем представлены общественности под видом лекарственных препаратов и ложно рекламировались как «безопасные и эффективные вакцины».
Лица, которые назначали, приобретали и/или вводили инъекции COVID-19 (мРНК), участвовали в военных преступлениях и/или геноциде (демоциде).
Ссылки и источники:
[1] https://en.m.wikipedia.org/wiki/List_of_U.S._biological_weapons_topics
[2] https://www.ncbi.nlm.nih.gov/books/NBK535870/
[3] https://sites.dartmouth.edu/dujs/2013/03/10/genetically-engineered-bioweapons-a-new-breed-of-weapons-for-modern-warfare/
[4] https://www.ncbi.nlm.nih.gov/books/NBK535887/
[5] https://irp.fas.org/threat/cbw/nextgen.pdf
[6] https://isgp-studies.com/jason-group-national-security-science
[7] https://www.ncbi.nlm.nih.gov/books/NBK535870/
[8] https://www.ema.europa.eu/en/human-regulatory-overview/research-and-development/compliance-research-and-development/good-manufacturing-practice/mutual-recognition-agreements-mra
[9] https://www.fda.gov/emergency-preparedness-and-response/about-mcmi/medical-countermeasures
[10] https://aspr.hhs.gov/legal/PREPact/pages/default.aspx
[11] https://www.fda.gov/regulatory-information/federal-food-drug-and-cosmetic-act-fdc-act/fdc-act-chapter-v-drugs-and-devices
[12] https://massie.house.gov/news/documentsingle.aspx?DocumentID=395737
[13] 21 USC § 360bbb-3(c)(2)(A)
[14] 21 USC § 360bbb3(c)(2)(B)
[15] 21 USC § 355(d)(2); См. также 42 USC § 262(a)(2)(RB) (биологический препарат одобрен только в том случае, если он действительно «…безопасен»).
[16] 21 USC 360bbb-3a(c).
[17] 21 USC § 360bbb-3(a)(1), (b).
[18] https://public-inspection.federalregister.gov/2024-29108.pdf
[19] https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32020R0521&from=EN
[20] https://www.bmj.com/content/372/bmj.n627
[21] https://dn721909.ca.archive.org/0/items/contract_03/Contract%203_text.pdf
[22] https://www.eppo.europa.eu/en/media/news/ongoing-eppo-investigation-acquisition-covid-19-vaccines-eu
[23] https://www.fda.gov/news-events/public-health-focus/expanded-access
[24] https://dn721909.ca.archive.org/0/items/contract_03/Contract%203_text.pdf
[25] https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q6a-specifications-test-procedures-and-acceptance-criteria-new-drug-substances-and-new-drug-products
[26] https://www.nature.com/articles/sigtrans20154
[27] https://pmc.ncbi.nlm.nih.gov/articles/PMC8067914/
[28] https://www.statnews.com/2017/01/10/moderna-trouble-mrna/
[29] https://thewashingtonstandard.com/moderna-a-company-in-need-of-a-hail-mary/
[30] https://www.govinfo.gov/content/pkg/PLAW-109publ417/pdf/PLAW-109publ417.pdf
[31] https://www.in.gov/dhs/files/FEMA-Fact-Sheet-COVID-Response-3.4.20.pdf
[32] См. “VRBPAC-10.22.20-Meeting-Presentation-COVID19-Vaccine-Development-Portfolio.pdf” в Приложении
[33] https://www.statnews.com/pharmalot/2020/09/28/pharmalittle-operation-warp-speed-is-more-army-than-science-jjs-covid-19-vaccine-moves-forward/
[34] См. “VRBPAC-10.22.20-Meeting-Presentation-COVID19-Vaccine-Development-Portfolio.pdf” в Приложении
[35] https://www.keionline.org/covid-contracts
[36] https://www.keionline.org/bn-2020-3
https://www.ati.org/
[38] https://www.medcbrn.org/current-members/
[39] https://www.keionline.org/misc-docs/DOD-ATI-Pfizer-Technical-Direction-Letter-OTA-W15QKN-16-9-1002-21July2020.pdf
[40] https://www.cdc.gov/vaccines/covid-19/vaccination-provider-support.html#6-23-22
[41] Правительство Нидерландов раскрыло секретное обязательство соблюдать цели НАТО по обеспечению устойчивости (новостная статья): https://deanderekrant.nl/kabinet-erkent-navo-is-de-baas/
Сегодняшняя работа: «Юнтвиллские дубы», акварель. Работы доступны здесь .




Я скачал этот документ для надежного хранения, поскольку доступ в интернет все больше ограничивается.
Я снова воспользуюсь этим, чтобы написать в правительство. На мои письма либо не отвечают, либо отвечают пренебрежительно — даже на те, которые отправлены заказным письмом, не подтверждают получение, нет никаких доказательств доставки.
Что тут скажешь? У нас нет верховенства закона. Наши правительственные чиновники — коллаборационисты и предатели.
Мой депутат не отвечает. Правительственные ведомства тоже. Вестминстер также не признает получения заказных писем. Вы говорите в пустоту. «Общественные расследования» существуют для того, чтобы установить исторические факты так, как им заблагорассудится.
Пандемия, как и любые связанные с ней дискуссии, безусловно, являются секретным вопросом безопасности.
Правительство сделает все возможное, чтобы отрицать причастность убийцы. Это потому, что оно хочет иметь возможность повторять это миллион раз. Все это держится в секрете, и никто не должен знать, что это происходит.